但单一国家或地区的临床数据 , 并不是阻碍信迪利单抗在美上市的唯一因素 。
按照相关监管法规 , 如果一款药物的相关数据满足美国人口和美国医疗实践 , FDA仍可采用“灵活的办法”来评估该药物的临床数据 。 享受“监管灵活性”的前提则包括:满足未被满足的临床需求;罕见病 , 难以开展全球多地区/中心临床试验;原创新药 。
对这些“灵活监管”要求 , 信迪利单抗也没有满足 。 一名委员在会上直言:“这项申请并没有解决一个未被满足的需求 , 我们已经有安全有效的治疗方法 , 在总体生存率上有改善 。 ”
此外 , ORIENT-11的试验设计不符合FDA的要求也成为讨论的焦点 。
有委员指出 , 在ORIENT-11启动时 , 一线转移性肺癌的护理标准已经发生了重大变化 , 即K药联合化疗在2018年获批用于美国的非鳞非小细胞肺癌治疗 , 而ORIENT-11研究选择培美曲塞联合铂类治疗作为对照组的临床试验设计已经“过时”了 。 换言之 , 信迪利单抗要在美国上市 , 需要跟其他有相同适应症的已上市PD-1药物作“头对头”试验 , 以证明自己对患者有“明显改善” , 而非仅仅证明自己的非劣性 。
此外 , ORIENT-11的设计终点也与美国现行肿瘤药一线治疗临床设计的主要终点不同 。
而之所以作出“过时”的临床试验设计 , FDA认为这是信达生物方面没有就试验设计等与其进行沟通咨询的结果 。 一名委员直指 , 信达和礼来方面是在试验初步结果出炉后才找到FDA , 而在此之前 , FDA甚至不知道试验正在进行中 。 在会议中 , FDA还罗列出了与信达/礼来沟通的时间轴 , 以说明信达/礼来在2018年8月直接启动了临床试验 , 但FDA在2020年4月才知晓这场试验的进行 。
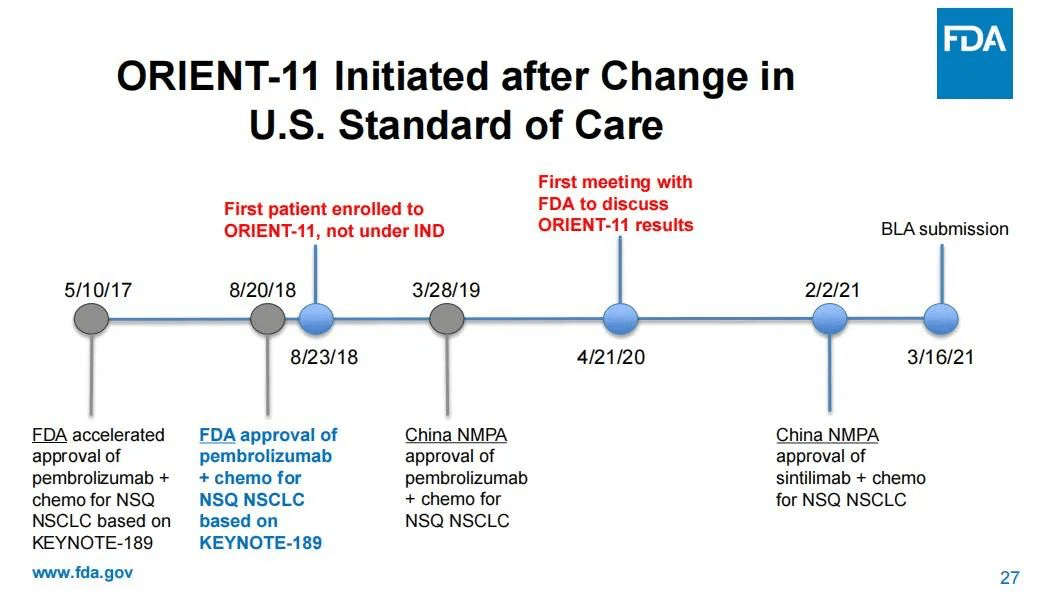
文章图片
图片来源:FDA审查资料截图
针对该质疑 , 信达及合作方礼来进行了解释 。 信达生物方面在接受媒体采访时表示:“不管在临床注册还是上市申报阶段 , 公司都和FDA保持了密切沟通 , 临床方案也获得了FDA认可 。 在2020年8月与FDA分别召开了pre-BLA关于临床和CMC的会议 , 对BLA递交资料的内容和形式进行了进一步的确认 , 我们并没有收到FDA对于单一国家数据临床申报的反对意见 , 或者要求必须是MRCT 。 ”
虽然对判定的细节尚有争议 , 但确定的是 , 信迪利单抗的美国上市节点还未到来 。 有市场人士预测 , 如果按照FDA的“头对头”及地区/中心要求补充临床试验 , 将是几亿美金和若干年的巨大投入 , 初步预估需要2000人 , 到2030年才会完成 。 即便是信达和礼来打算继续投入 , 也要考虑投入与回报三思而行 。
关键是填补临床需求:不应为权宜之计牺牲质量
不难发现 , “单一国家/地区临床试验数据”、“不满足未被满足的临床需求”、“未与FDA充分沟通”是造成信达生物此次出海暂缓的主要原因 。
其中 , “不满足未被满足的临床需求”成为首当其冲的关键因素 。 东吴证券医药行业首席分析师朱国广认为 , 尽管上述FDA ODAC会议花费了大量时间讨论ORIENT-11的临床终点、对照药物和人种多样性等 , 但信迪利单抗被拒绝的核心原因仍在于“临床需求的紧迫性不够” 。 在一个有类似药物的领域 , FDA势必用更高标准要求临床质量 。
信迪利单抗此次申请上市的是PD-1单抗的非鳞非小细胞肺癌适应症 , 在美国市场已经有“O药”“K药”“T药”以及西米普利单抗等治疗药物上市 。 以目前非鳞非小细胞肺癌的标准疗法“K药”为例 , 其于2014年获批上市 , 2021年实现全球销售收入171.86亿美元 , 仅次于常年霸占销售排行榜第一的“药王”阿达木单抗 。
- 市场|找到衰老开关?中国科学家:母乳中关键分子,可增强老年机能
- 美国人养牛为什么要在牛身上开个洞?我国为什么没有推广此法?
- 本文转自:农村大众唐乐观光合作社的农产品展厅里|这个村办蔬菜市场交易量交易金额双过亿,收益村民共享
- 疫苗|美国FDA考虑秋季批准第四剂新冠疫苗
- 美国医学会|好好睡觉能瘦身
- 死亡|美国新增确诊19202例、死亡383例
- 美国杂志对汉服的科普,又让韩国人破防了?
- 美国队|冬奥村里的“社区诊所”:中医科号源供不应求
- 全套疫苗也没用?美国传来一则紧急噩耗,担心的事还是发生
- 破罐子破摔!3亿美国人主动“躺平”,向来淡定的白宫有点急了